Visualisation scientifique 3D avec Streamlit, PyVista et Ipyvolume (partie 1)
Nous faisons beaucoup d’analyses exploratoires de données, avec leur lot de visualisations diverses. On en fait plus rarement en 3D, étant moins confrontés à ce type de données ; nous allons voir qu’il n’est pourtant pas très compliqué de réaliser des rendus simples mais jolis à voir et à manipuler, et la 3D fait toujours son petit effet. Nous vous proposons une paire d’articles détaillant la réalisation d’une application de visualisation de structures cristallines, prétexte au mariage de trois librairies : Streamlit pour l’application interactive, et le couple PyVista + Ipyvolume pour les rendus en 3D. Nous resterons donc dans le monde Python bien connu des data scientists.
Nous avons découpé le discours en deux parties pour en facilier la lecture:
- la première partie, celle-ci, présente le cas d’application visé ainsi que les visualisations 3D au moyen de PyVista et Ipyvolume ;
- la seconde partie s’intéressera à l’application Streamlit complète, intégrant donc les deux librairies précédentes
Vous trouverez à cette adresse le code source complet de l’application décrite dans l’article, avec quelques données fictives.
Les structures cristallines
Avant de rentrer dans la technique, parlons un peu des données que nous allons manipuler. Nous sommes partis d’un jeu de données publié sur la plateforme de data science Kaggle : Nomad2018. La partie du jeu de données qui nous intéresse est une série de fichiers .xyz qui décrivent l’organisation d’atomes dans des cristaux (un fichier par composé). Le format .xyz utilisé est très facile à lire, en voici un exemple :
# Un composé cristallin fictif, qui à moins d'un hasard extraordinaire
# n'existe pas dans la nature.
#
# Toutes les grandeurs sont sous-entendues en angströms (Å)
# Les 3 vecteurs formant les côtés de la maille cristalline
# Ils ne sont pas forcément orthogonaux !
lattice_vector 1.234 -0.123 0
lattice_vector 0.456 3.142 0.178
lattice_vector -0.333 0.456 2.718
# Les coordonnées des centres et les natures des atomes qui constituent la maille
# (en nombre arbitraire)
# Dans ce dataset, il n'y a que 4 espèces : aluminium (Al), indium (In), gallium (Ga), oxygène (O)
atom 0.11 0.12 0.13 Al
atom 0.21 0.22 0.23 In
atom 0.31 0.32 0.33 In
atom 0.41 0.42 0.43 Ga
atom 0.51 0.52 0.53 O
atom 0.61 0.62 0.63 O
En l’occurrence les composés décrits sont des semi-conducteurs, et le but du concours était d’en prédire les propriétés à partir de leur structure atomique. Nous nous contenterons de les afficher !
Pour se procurer les fichiers du dataset Nomad2018, il faut s’inscrire sur la plateforme, se déclarer comme participant à au concours (terminé depuis longtemps), et télécharger les fichiers ZIP qui le constituent. Des fichiers fictifs sont aussi fournis à titre d’exemple avec le code de l’application, plus pratiques pour vérifier les calculs.
Pour rappel, un cristal est un matériau composé d’atomes organisés en motifs qui se répètent dans l’espace. Le motif élémentaire est la maille du cristal, dont les atomes sont listés dans le fichier .xyz sous forme d’entrées atom. Les vecteurs de la maille, lattice_vector, représentent les translations élémentaires qui génèrent les copies de la maille. Chaque atome A de la maille est ainsi infiniment répliqué dans 3 directions, donnant des atomes Ak,l,m = A + k__v__1 + l__v__2 + m__v__3 ; k, l, m ∈ ℤ (nous utilisons une notation vectorielle, A est vu comme un vecteur dont les coordonnées sont le centre de l’atome).
Dans le code de notre application, c’est le module io/xyz.py qui est chargé de lire les fichiers .xyz. Pour un fichier donné, il renvoie des tableaux NumPy avec les informations contenues dedans, avec un petit plus : chaque atome se voit attribuer un rayon, dit de Van der Waals, qui est une estimation de la taille de la couche électronique externe de l’atome seul, supposé sphérique. On trouve ces valeurs dans des tables. Bien sûr, dans un semiconducteur les électrons sont partiellement délocalisés, et les atomes ne sont pas des sphères immobiles, mais il est difficile de faire mieux sans se lancer dans des simulations quantiques compliquées, ce dont l’auteur de cet article est à son grand regret bien incapable…
A partir de ces données, que pouvons-nous visualiser ? Voici ce que notre application va proposer :
- une vue de la structure atomique dans l’espace, avec les atomes et les vecteurs de la maille
- une vue d’un “champ de potentiel” dans une région de l’espace, tenant compte de la répétition de la maille. Nous mettons des guillemets à “champ de potentiel” car celui que nous allons implémenter n’a pas de sens physique, mais rien n’empêche d’en calculer un !
Les visualisations
Dans cette partie nous détaillons l’implémentation de chacune de ces visualisations. Nous avons choisi deux librairies, une pour chaque, ce qui peut paraître étrange. Cela s’explique par la facilité de prise en main de l’une et de l’autre au regard de notre ambition… nous allons bien sûr expliquer ces choix, qui sont d’ordre tactique.
Vue de la structure atomique
Nous souhaitons représenter les atomes sous forme de sphères, centrées aux positions des atomes, et avec un rayon proportionnel à celui des atomes réels. Nous voulons aussi voir les vecteurs qui constituent la maille, comme on représente une base de l’espace (ce qu’ils sont en réalité).
En image:
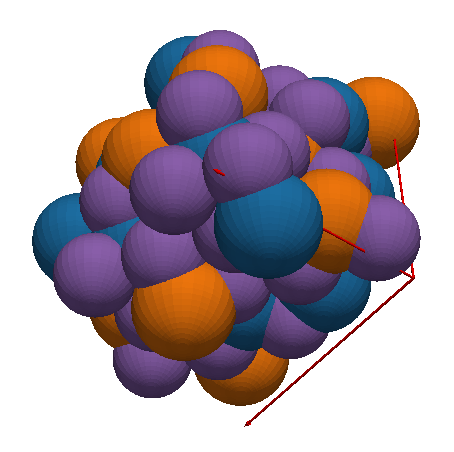
Dans l’exemple ci-dessus, nous voyons 80 atomes d’aluminium (bleu), de gallium (orange) et d’oxygène (violet). Il n’y a pas d’indium dans ce composé. Les vecteurs de la maille sont en rouge. En termes de représentation, chacun de ces objets – sphères et flèches – sont ce qu’on appelle des maillages ou meshes, composés de polygones élémentaires. Les surfaces arrondies sont simulées par la juxtaposition de rectangles ou de triangles, plus simples à rendre et qui font bien illusion grâce à l’éclairage simulé.
Pour ce type de rendu, nous avons choisi PyVista, qui propose des primitives géométriques de haut niveau, comme les sphères et les flèches justement. Il suffit de les assembler pour disposer d’une visualisation complète en quelques lignes.
Voici le morceau de code qui effectue le rendu de la structure :
import pyvista as pv
plotter = pv.Plotter(notebook=True)
plotter.set_background('white')
# Des valeurs raisonnables compte tenu des échelles en jeu
axes_properties = {
'scale': 'auto',
'tip_length': .03,
'tip_radius': .01,
'shaft_radius': .005
}
# vectors contient nos 3 vecteurs de maille
for vector in vectors:
plotter.add_mesh(pv.Arrow(direction=vector, axes_properties), color='red')
# atoms est un tuple (kinds, centers, radii)
# ATOM_COLORS attribue des couleurs aux types d'atomes
for kind, center, radius in zip(*atoms):
plotter.add_mesh(
pv.Sphere(radius, center),
color=ATOM_COLORS[color_scheme][kind],
)
# Evite que PyVista ne masque les objets trop "éloignés", en calant la région
# visible sur celle occupée par les objets
plotter.reset_camera_clipping_range()
# Récupère le contenu de la scène sous forme de widget IPython
# 'pythreejs' est un backend de rendu JS pour PyVista, voir la doc pour les alternatives
scene = plotter.show(jupyter_backend='pythreejs', return_viewer=True)
Le tuple atoms, qui contient les natures, coordonnées et rayons des atomes, est issu directement du fichier .xyz. Il en est de même pour les 3 vecteurs de la maille (tableau vectors).
Cela nous fait donc plusieurs objets – plusieurs maillages – cumulés sur le même graphique. PyVista nous permet de les composer, tout comme Matplotlib lorsqu’on appelle plusieurs primitives avant de faire un plt.show(). Nous verrons plus loin que faire de la scène ainsi générée, pour l’afficher dans la page Streamlit.
Une fois le graphique affiché, il sera possible de le faire tourner dans l’espace avec la souris pour regarder notre cristal sous toutes les coutures. Il est d’ailleurs assez fascinant de le manipuler ! On a bien mérité un café.
Le champ de potentiel
Cette vue est très différente de la précédente, la donnée représentée étant un champ scalaire, i.e. une valeur réelle associée à chaque point de l’espace. Le rendu fait comme si chacun de ces points, ou voxels, était un cube élémentaire d’une densité donnée, et par transparence on a un aperçu de tout le volume.
Voici une capture sur une région étendue, qui met en évidence la périodicité du champ dans l’espace :
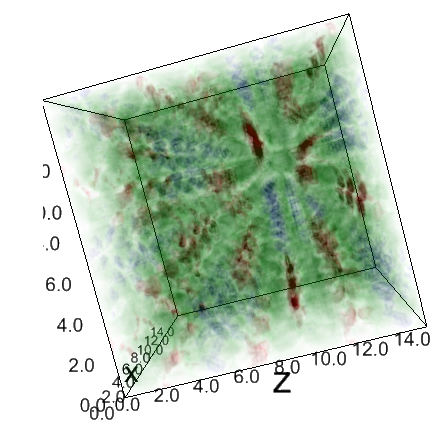
Nous aimerions bien utiliser PyVista pour faire cette vue. Il y a d’ailleurs une fonction plotter.add_volume() qui fait exactement cela mais… elle ne marche pas dans notre cas. Ou du moins, on n’a pas réussi, pour des raisons qui seront expliquées dans le deuxième article. Plutôt que de nous acharner, nous avons pris une autre option, avec la librairie Ipyvolume.
Comme son nom l’indique, Ipyvolume a pour vocation de faciliter l’affichage interactif de volumes. La primitive qui permet de faire un rendu volumétrique s’appelle volshow(). Son invocation ressemble à ceci :
import ipyvolume as ipv
# voxels : tableau NxNxN avec les scalaires
à représenter
# extent : l'étendue du cube à représenter (un peu comme les limites du scatter plot)
# tf : fonction de transfert (voir ci-après)
ipv.volshow(voxels, extent= * 3, tf=tf)
Nous avons nommé le premier paramètre voxels, car c’est bien de cela qu’il s’agit : la généralisation en 3D de la notion de pixels. On voit, sur l’image donnée en exemple, que le résultat est ouaté et bigarré, alors que chaque voxel porte un simple scalaire. C’est la fonction de transfert (transfer function) qui fait cette conversion en fausses couleurs ; si nous n’en fournissons pas (tf = None), une fonction par défaut est appliquée et des contrôles supplémentaires apparaissent automatiquement pour en ajuster les paramètres :
La fonction de transfert est en fait une interpolation linéaire par morceaux entre les valeurs scalaires des données fournies (entre voxels.min() et voxels.max()), et un gradient de couleurs RGBA (il y a aussi un lissage automatique des faces, qui contribue à l’aspect nébuleux). La fonction de transfert par défaut possède 3 noeuds d’interpolation mobiles (en plus des extrêmes), les levels ci-dessus, correspondant à des couleurs rouge, verte et bleue respectivement, et dont les opacités se règlent avec les curseurs du dessous. Elle a l’avantage de laisser l’utilisateur choisir les seuils lorsque la visualisation est complexe et dense ; certains traits visuels ne sont pas faciles à mettre en évidence automatiquement. Dans l’application, nous laisserons le choix entre cette fonction par défaut, et une plus simple (une rampe d’opacité monochrome), plus “naturelle” pour représenter des densités, mais moins flexible.
En attendant la suite
Afficher des données sous deux formes différentes, des sphères et des volumes translucides, s’avère simple. Les morceaux de code que nous avons vus sont ceux que l’on utiliserait pour embarquer les visualisations dans un notebook Jupyter, qui est la cible de choix des PyVista et surtout d’Ipyvolume.
Toutefois, il reste un peu de travail pour les intégrer à une application proprement dite, avec Streamlit donc, et cela fait l’objet du deuxième article.
Vous trouverez en annexe une explication rapide du calcul du champ de potentiel, ou ce qui en tient lieu, utilisé dans le code.
Annexe : le calcul de "potentiel"
Attention, comme annoncé ce n’est pas réaliste, comme le serait un potentiel électrostatique tenant compte de la distribution des électrons dans le cristal. A des fins d’illustration, nous avons choisi une solution de facilité : le “potentiel” en un point de l’espace est le nombre d’atomes qui contiennent ce point. Les atomes sont considérés comme des sphères de taille prévisible (leur rayon de Van der Waals), qui se recouvrent parfois. En l’occurrence, les points où le potentiel sera évalué sont les centres des voxels que nous souhaitons représenter avec ipv.volshow().
Voici une illustration du principe en 2 dimensions, une représentation que nous utiliserons dans la suite par simplicité. Le nombre dans chaque case (chaque voxel) est donc le nombre de sphères qui passent par son centre.
Compter les atomes présents un un point de la maille unitaire, c’est facile, ce sont des sphères dont nous connaissons les centres et les rayons. Voici notre implémentation, qui compte les sphères couvrant un point M<i>cell</i> (voir preprocessing/overlaps.py) :
def lookup_voxel(M_cell: np.ndarray, centers: np.ndarray, squared_radii: np.ndarray) -> float:
squared_distances = ((M_cell - centers)2).sum(axis=1)
return (squared_distances <= squared_radii).sum()
La complexité vient du fait que notre cristal est périodique, nous retrouvons des atomes dans tout l’espace. Le fichier .xyz ne contient que la maille élémentaire, bien sûr, et nous ne savons pas a priori combien de fois il faut la recopier. Cela nécessiterait de simuler les atomes des autres mailles, autant de fois qu’il faut pour couvrir la région à représenter, et ainsi traiter le cas où Mcell n’est pas dans la cellule. Ce ne serait pas très “fin” comme approche, on peut faire mieux.
Prenons le problème dans l’autre sens : pouvons-nous “ramener” un point quelconque M de l’espace dans cette maille unitaire ? En quelque sorte, nous voudrions Mcell = M mod “la maille unitaire”. C’est possible avec un changement de coordonnées ; les vecteurs de la maille, récupérés du fichier .xyz, forment une matrice de passage entre l’espace réel et celui du cristal. Quelques manipulations d’algèbre linéaire, domaine où NumPy excelle, vont faire le travail.
Dans le code, le paramètre vectors de la fonction get_voxels(), issu directement de la lecture du fichier, est structuré ainsi :
C’est presque la matrice de passage de la base canonique à celle de la maille, il faut juste la transposer : vectorsT. En appliquant l’inverse de cette matrice à un point M, on obtient un vecteur que nous appelons Mlattice = (vectorsT)-1 M et qui contient les coordonnées de ce point dans le réseau du cristal. Dans cette base, le coin extrême de la maille élémentaire a pour coordonnées (1,1,1). Dans le cas d’un vecteur Mlattice quelconque, prendre la partie fractionnaire de ses coordonnées revient à les ramener à l’intérieur de la maille unitaire, notre objectif ; reste à remultiplier par la matrice de passage pour retrouver nos coordonnées réelles, dont nous avons besoin pour appeler lookup_voxel(). Pour résumer : Mcell = vectorsT ⎣ (vectorsT)-1 M ⎦.
Il reste un détail à régler. Certains atomes “débordent” de la maille à cause de leur rayon. Il est donc possible de retrouver, dans la maille unitaire, des portions d’atomes voisins répliqués, que nous devons comptabiliser dans le “potentiel”. Voici un exemple avec le gros atome qui déborde de la maille d’à côté :
La fonction lookup_voxel() ne fait pas ce travail ; nous devons alors lui passer une liste d’atomes et de rayons “augmentés” par une copie de la maille dans chaque direction. Nous voulions échapper à la réplication explicite des atomes, moralité il faut quand même le faire. Nous nous limitons toutefois à une seule réplication dans chaque direction, en faisant l’hypothèse, réaliste ici, qu’un atome n’ira pas au-delà des mailles immédiatement voisines.

Cela fait tout de même une belle augmentation des calculs ; pour 80 atomes de base, par exemple, l’augmentation conduit à un total révisé de 3^3 x 80 = 2160 atomes fictifs, autant de sphères à examiner pour chaque point de notre tableau de voxels… c’est toujours mieux que de devoir répliquer explicitement la maille pour couvrir une région arbitraire de l’espace, puisque le nombre de sphères à ajouter varie avec le cube de la taille de la région. Pour ce qui est de l’implémentation, la fonction lookup_voxel() utilise pleinement la vectorisation des calculs avec NumPy, nous avons au moins évité une boucle Python sur les atomes.